|
Salud Dental
|
Fisiología de la hemostasia
La acción quirúrgica, dado que necesariamente secciona y lesiona los tejidos orgánicos, produce soluciones de continuidad en el sistema vascular, unas veces a nivel de la macrocirculación y siempre en la microcirculación (arteriolas, capilares y vénulas).
La consecuencia es la hemorragia operatoria, es decir, el flujo de la sangre fuera del sistema vascular, sea arterial o venoso, y los fenómenos generales consiguientes a esas hemorragias, cuando sobrepasan cierto límite sin ser controladas, son ya conocidos: hipovolemia e hipoperfusión de los tejidos que puede llegar hasta el estado de shock hemorrágico constituido.
De estas consideraciones iniciales se deduce la gran importancia que tiene para el cirujano dental y maxilofacial el conocimiento preciso de la hemostasia en sentido muy amplio, es decir, del conjunto de procesos biológicos y de procedimientos técnicos quirúrgicos que sirven para detener y controlar la hemorragia.
La hemostasia puede ser considerada en su aspecto espontáneo o natural o bien desde el punto de vista de la técnica quirúrgica.
La hemostasia espontánea o natural puede ser definida como el conjunto de procesos biológicos, precisamente integrados, cuya finalidad es conseguir que la sangre se mantenga dentro del sistema vascular (hemostasia natural estática), obturando las soluciones de continuidad que se produzcan en los vasos (hemostasia natural correctora). La hemostasia quirúrgica agrupa todos los procedimientos técnicos que el cirujano emplea para controlar la hemorragia que se produce accidentalmente o durante el acto operatorio.
En toda intervención quirúrgica para dominar la hemorragia son precisas las dos formas de hemostasia, ya que mientras las técnicas de la hemostasia quirúrgica (ligaduras, coagulación térmica, presión mantenida, etc.) cierran los vasos macroscópicos, la hemostasia natural o espontánea detiene, de modo preferente, la hemorragia que se produce en la extensísima microcirculación lesionada en el campo operatorio.
La hemostasia natural tiende a conseguir la formación de un coágulo resistente que cierre la solución de continuidad y detenga la salida de la sangre. La hemostasia efectiva depende de unas complejas interacciones entre:
- pared vascular
- plaquetas
- proteínas plasmáticas implicadas en la coagulación (factores plasmáticos)
Ante una lesión vascular, se producen sucesivamente tres fases:
- Fase vascular
- Fase plaquetaria
- Fase de la coagulación plasmática
Si bien esta distinción sirve a los propósitos de la comprensión y exposición didáctica, todo el proceso debe ser considerado como una serie de secuencias íntimamente relacionadas e integradas, constituyendo la tríada de la hemostasia.
A esto se añade un complejo sistema de inhibidores fisiológicos y mecanismos de control que permiten delimitar cualquier activación excesiva o inadecuada del proceso hemostático.
Fase vascular
Producida la solución de continuidad en la pared de un vaso, se inicia rápidamente (en décimas de segundo) una respuesta vasoconstrictora, debida en parte a reflejos nerviosos locales (axónicos) y espinales, y también a la acción de ciertas aminas vasoactivas liberadas por la acción traumática, entre ellas la serotonina.
Esta respuesta vasoconstrictora cumple dos finalidades en la hemostasia: por una parte disminuya la pérdida de sangre, gracias al cierre del vaso lesionado y por otra inicia la segunda fase, plaquetaria, facilitando la adhesión de las plaquetas. En esta acción facilitadora influye, probablemente, una alterqación en la carga eléctrica de la íntima (haciéndola positiva) y también la exposición de las fibras colágenas de la pared vascular lesionada, denudada de su endotelio.
Las conexiones entre la fase vascular y la plaquetaria se acentúan si recordamos que las plaquetas poseen también una función protectora del endotelio, caso por medio de su incorporación al citoplasma de las células endoteliales; precisamente en los estados trombopénicos se suelen presentar lesiones endoteliales. Existe una unidad funcional endotelio-plaquetas que relaciona íntimamente las dos primeras fases de la hemostasia.
Por otro lado, la síntesis de la sustancia intercelular del endotelio, precisa de la vitamina C, lo que explica las manifestaciones purpúricas del escorbuto.
Fase plaquetaria
En esta fase se realiza la constitución del trombo o clavo plaquetario ("cabeza blanca" del trombo definitivo), al mismo tiempo que en la agregación plaquetaria tiene lugar la concentración de una gran cantidad de factores necesarios para la tercera fase de la coagulación plasmática.
Las plaquetas son los elementos formes más pequeños de la sangre circulante (un tercio del tamaño de los hematíes) de forma discoide y sin núcleo. Son producidas por la fragmentación del citoplasma de los megacariocitos de la médula ósea y acaso también de los situados en el pulmón. Los megacariocitos son las células más grandes de la médula ósea. Derivan de la célula madre pluripotencial que, bajo el influjo de hormonas trombopoyéticas o "trombopoyetinas", son inducidas en la línea megacariocítica.
El megacariocito es la única célula de la médula ósea que tiene capacidad de reproducir su DNA sin sufrir división celular (endocitosis). Se ha estimado que un megacariocito da lugar a 1.000 plaquetas. La secuencia madurativa dura cuatro a cinco días.
Siendo las plaquetas de forma aproximadamente esférica, su diámetro varía entre 2 y 4 micras, con 7 a 8 micras cúbicas de volumen.
Su membrana protoplásmica, de estructura lipoproteica, con un espesor aproximado de 20 a 30 milimicras, es rica en la enzima ATP-asa (adenosintrifosfatasa). Alrededor de esta membrana se dispone una "atmósfera plasmática periplaquetaria" rica en factores de la coagulación.
La cantidad normal de plaquetas oscila entre 150.000 y 300.000 por mm3. Se encuentran acumuladas en el bazo y en el pulmón y son destruídas en el sistema reticuloendotelial (hígado y bazo). No se encuentran plaquetas en la linfa del conducto torácico. La vida media de las plaquetas oscila entre 9 y 11 días.
Las funciones de las plaquetas en la fase plaquetaria trascienden de este estadio para aportar mecanismos importantes tanto a la primera fase, vascular, como a la siguiente, plasmática. Por estas razones, las actividades funcionales de las plaquetas han sido divididas en la (Tabla 1):
- Funciones dinámicas, correspondientes a la adhesividad y a la agregación plaquetaria, la metamorfosis viscosa, la función trombodinámica y la función retráctil.
- Funciones plasmáticas, cumplidas mediante la liberación de factores para la tercera fase (coagulación) e incluso para la primera fase (serotonina con acción vasoconstrictora).
|
Tabla 1: Funciones plaquetarias |
|
| 1. Funciones dinámicas | Adhesividad Agregación Metamorfosis viscosa Función trombo dinámica Función retráctil |
| 2. Funciones plasmáticas | Liberación de factor plaquetario 3 Liberación de
factor 2 (acelerador de la trombina) Liberación de factor 4 (factor antiheparina) |
Funciones dinámicas
El acontecimiento inicial de la hemostasia es la adherencia de las plaquetas a las fibras de colágeno (especialmente el colágeno de tipo III) y a otras materias fibrilares del subendotelio. La adhesión requiere que la plaqueta forme una unión estable con la superficie del vaso, y esto se hace a través de la participación de al menos dos cofactores: el factor de von Willebrand (sintetizado por las células endoteliales de la pared vascular) y la fibroconectina (sintetizado por el subendotelio vascular). Esta unión es dependiente de la activación plaquetar y está mediatizada por la presencia de Ca++. Las plaquetas forman al principio una monocapa.
Antes de la adherencia y, sobre todo, después, se desarrolla otro fenómeno plaquetario denominado agregación, en virtud del cual se adhieren entre sí, formando el trombo blanco. Tras la agregación reversible tiene lugar la agregación irreversible y la metamorfosis viscosa, proceso durante el cual las plaquetas agregadas pierden sus gránulos, emiten seudópodos y se transforman en una masa viscosa sin contornos individuales, por lisis de sus membranas. La fase de agregación reversible puede anularse sustituyendo el calcio mediante la adición de sustancias descalcificantes como el citrato y el oxalato.
El trombo blanco constituido, obturando la solución de continuidad, es la respuesta primaria o provisional en el mecanismo de la hemostasia espontánea o natural. Su duración suele ser de tres a cuatro horas, hasta que se produce su lisis.
Funciones plasmáticas
Para su intervención en la tercera fase, las plaquetas disponen de los siguientes factores:
| Factor 1 | Similar al factor V de la coagulación. |
| Factor 2 | Dotado de actividad fibrinoplástica, acelera la conversión del fibrinógeno en fibrina. |
| Factor 3 | Es el factor plaquetario más importante para la coagulación. Está constituido por una fosfolipoproteinemia y acelera la formación de la tromboquinasa o tromboplastina. |
| Factor 4 | Se trata de una antiheparina que neutraliza a ésta y a sustancias con efecto heparínico como el dextrano. |
| Trombastenina | Proteína contráctil que interviene en la retracción del coágulo. |
Los coágulos preparados a partir de plasma sin plaquetas se contraen en menor grado que los normales (plasma con plaquetas). La retracción del coágulo producida por la acción plaquetaria se estima que corresponde a un 50%. Esta proteína contráctil responsable de esta actividad plaquetaria es parecida a la actinomiosina del músculo y para su acción requiere la presencia de calcio, glucosa, ATP y un cofactor no determinado.
La función trombodinámica de las plaquetas es muy importante para la estructuración definitiva del coágulo de fibrina (sinéresis) transformando las fibras largas y gruesas en otras finas y cortas en disposición tridimensional. El trazado del aparato tromboelastográfico, aumentando la separación de las líneas de su trazado (de 20 nm en ausencia de plaquetas a 60 nm en su presencia) hace objetiva esta función trombodinámica.
Fase de la coagulación plasmática
En este estadio del proceso de la hemostasia se distinguen, a su vez, dos periodos: primero, la formación del coágulo y después su lisis. El resultado es que una proteína soluble en el plasma, el fibrinógeno, se convierte en una proteína insoluble, la fibrina. Esta reacción es catabolizada por una enzima, la trombina. Esta no está presente en el plasma o la sangre circulante, pero sí su precursor inerte, la protrombina.
La hipótesis de "cascada" introdujo el concepto de que los factores de coagulación existirían de una forma "inactiva" o procoagulante, y de una forma "activa". La forma activa de un factor activaría especificamente el siguiente de una forma secuencial, dando lugar a la llamada "cascada". El proceso de activación para la mayoría de los factores se lleva a cabo por la "división" de una pequeña parte de la forma inactiva.
La mayoría de los factores activados son serina-proteasas, las cuales son una familia de enzimas proteolíticas con una serina en su centro activo. Los factores serina-proteasa tienen un alto grado de especificidad en el sustrato. Son excepciones el factor V, el factor VIII y el fibrinógeno.
En el siguiente esquema, muy simplificado, se encuentra una primera introducción a las secuencias de la coagulación plasmática; en él se distinguen tres estadios:
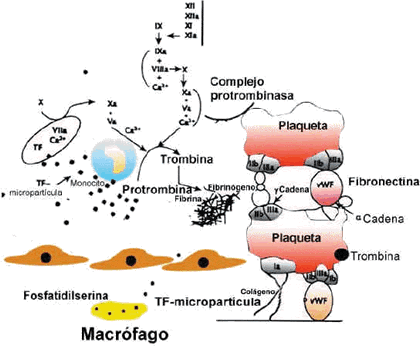 |
- En el primero se alcanza la formación de la tromboquinasa o tromboplastina.
- En el segundo la formación de la trombina.
- En el tercero la transformación del fibrinógeno en fibrina.
La trombina, una enzima proteolítica, es pues el factor clave en el proceso que se inicia en la fase anterior a la agregación plaquetaria, comienza la formación de la fibrina e incluso, como después veremos, activa la fibrinasa (factor XIII), enzima que actuando dentro de la molécula de fibrina ya formada, consigue una estructura más resistente.
Como quiera que en la exposición del proceso de la coagulación vamos a utilizar la moderna terminología, conviene que comparemos ésta con las denominaciones clásicas (Tabla 2).
| Tabla 2: Terminología de los factores de la coagulación | ||
| número romano | nombre | sinónimo |
| I |
Fibrinógeno |
|
| II |
Protrombina |
|
| III |
Tromboplastina |
Tromboquinasa |
| IV |
Calcio |
|
| V |
Proacelerina |
Factor lábil, globulina acelerada (Ac-G) |
| VI |
Igual que el factor V (este término se utiliza generalmente) |
|
| VII |
Proconvertina |
Factor estable, acelerador de la conversión de la protrombina del suero (SPCA) |
| VIII |
Globulina antihemofílica (AHG) |
Factor antihemofílico A |
| IX |
Componente de la tromboplastina del plasma (PTC) |
Factor Christmas, factor antihemofílico B |
| X |
Factor Stuart-Prover |
Autoprotrombina C |
| XI |
Antecedente de la tromboplastina del plasma (PTA) |
Factor antihemofílico C |
| XII |
Factor Hageman |
Factor contacto, factor cristal ("glass factor") |
| XIII |
Factor estabilizador de la fibrina |
Fibrinasa, factor Laki-Lorand |
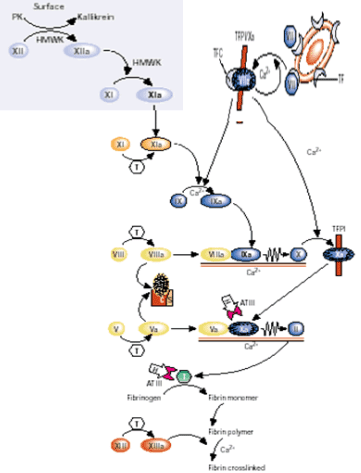
Clásicamente, se dice que la activación de la protrombina se podía hacer por dos vías o sistemas: sistema intríseco y sistema extrínseco. Veamos las diferencias.
Los términos intrínseco y extrínseco se refieren a la formación del coágulo dentro o fuera del sistema vascular. El sistema intrínseco es relativamente lento, y el extrínseco, más rápido. En ambos, la vía final es la conversión de protrombina en trombina, enzima activa que actúa sobre el fibrinógeno como sustrato.
Por definición, la activación intrínseca supone que la sangre no ha salido fuera de los vasos poniéndose en contacto con los tejidos perivasculares. El contacto anómalo de la sangre se produce dentro de los vasos (placas de ateroma en una arteria, próteis vasculares), o con sangre extraída de los vasos y depositada en un tubo de cristal (superficie humedecible en contraste con el carácter no humedecible del endotelio normal).
Este contacto anómalo inicia la vía intrínseca de la transformación de protrombina en trombina (cada molécula de protrombina se escinde en dos de trombina), mediante la activación del factor XII (factor Hageman).
La activación extrínseca se produce cuando la sangre se pone en contacto con los tejidos perivasculares lesionados y material procedente de estos tejidos penetra en la circulación (tromboplastina de los tejidos).
Esta vía comienza con la activación del factor VII por la tromboplastina de los tejidos en presencia de calcio, con lo que se evitan los cuatro primeros pasos de la coagulación cuando ésa se hace por medio de la activación extrínseca.
La distinción entre las dos vías de coagulación parece un poco arbitraria. Se ha comprobado que el factor VII se puede activar por el factor XIIa y por el IXa. A su vez, el factor tisular, el factor VIIa y el factor Xa, pueden activar al factor IX. El factor tisular funciona como un cofactor en la activación ver esquema inferior.
 |
La conversión del fribrinógeno (factor I) en fibrina es una reacción compleja: la trombina divide la molécula de fibrinógeno a nivel de unos enlaces específicos (de argininaglicina) liberando dos péptidos (fibrinopéptidos A y B), siendo uno de éstos una sustancia vasoactiva. La molécula proteica que resulta de esta escisión se polimeriza para formar largos agregados moleculares unidos por enlaces de hidrógeno.
En una primera fase, la fibrina formada es soluble en urea puesto que esta sustancia es capaz de romper los enlaches de hidrógeno; por esta razón se denomina fibrina soluble. En una segunda fase, mediante la actividad del factor XIII, llamado también fibrinasa Ver esquema, que es activado a su vez por la trombina, se producen dentro de la molécula de fibrina enlaces covalentes de disulfuro, con lo que se consigue una mayor estabilidad de su estructura: es la fibrina insoluble. Los pacientes con déficit de fibrinasa pueden sufrir hemorragias postoperatorias y presentar dificultades en la cicatrización de sus heridas.
Factores de coagulación
Han sido reconocidas doce proteínas (tabla 3). Las vamos a dividir en varios grupos funcionales:
- Vitamina K dependientes (factor IX, factor X, factor II y factor VII)
- Cofactores (factor V y factor VIII)
- Activación del sistema "contacto"
- Fibrinógeno y factor XIII (relacionados con la fibrina)
| Tabla 3: Factores de coagulación | ||
| grupos | factores de cogulación | lugar de síntesis |
| Factores vitamina K dependientes | II VII IX X |
Hígado (hepatocito) Hígado (hepaHemorragia. Hemostasia. Coagulación sanguínea. Trasfusiones.ito) Hígado (hepatocito) Hígado (hepatocito) |
| Cofactores | V VIII: C |
Hígado, plaqueta, células endoteliales Células endoteliales |
| Activadores del sistema de contacto | XI XII Prekalicreína Kininógeno |
Hígado (?) Hígado (?) Hígado (?) Hígado (?) |
| Fibrino-formación | Fibrinógeno XIII |
Hígado (hepatocito) Hígado, plaqueta(?) |
Factores vitamina K dependientes
La activación de estos factores depende de un adecuado suplemento de vitamina K, la cual viene de la dieta y, una pequeña proporción, de la síntesis bacteriana en el tracto gastro-intestinal. Los factores X, IX, II y VII sintetizados en ausencia de esta vitamina, son los llamados PIVKAS (proteínas inducidas por ausencia o antagonistas de la vitamina K); estas proteínas son inactivas y, para ser biológicamente activas, necesitan la "carboxilación" de los ácidos glutámicos residuales. La carboxilación ocurre en la posición g , conduciendo al ácido g –carboxil glutámico. Los antagonistas de la vitamina K (dicumarínicos) inhiben esta carboxilación y el resultado es un impedimento en la unión a los fosfolípidos en presencia de Ca.
Cofactores
El factor V y el factor VIII circulan en el plasma como precursores de cofactores, biológicamente inactivos. Siguiendo la activación, el factor V activado sirve como cofactor no enzimático para el factor X activado en el complejo "protrombinasa" y el factor VIII, como cofactor en la activación del factor X mediatizada por el factor IX activado.
Activadores de "contacto"
Los factores XII, XI, prekalikreína y kininógeno de alto peso molecular, están implicados en la activación del sistema intrínseco de coagulación cuando el plasma sanguíneo se pone en contacto con superficies o sustancias cargadas negativamente, como el vidrio, kaolín, celite, ácido elágico, etc. El factor XII, XI y prekalikreína son zimógenos de serina-proteasas. El kininógeno de alto peso molecular es un cofactor no enzimático para estas reacciones. Las reacciones de contacto, además de estar implicadas en la coagulación, se unen a otros sistemas proteolíticos plasmáticos:
- La kalikreína es capaz de liberar "kininas" vasoactivas dede el kininógeno.
- Activa el plasminógeno.
- Activa el C1.
Fibrinógeno y factor XIII
Ambos está relacionados con la formación de fibrina, por la actuación de la trombina. El fibrinógeno es uno de los mayores constituyentes del plasma. Circula entre dos fuerzas, la trombina en la formación del coágulo y la plasmina implicada en su disolución.
Cuando la trombina actúa enzimáticamente sobre él, "divide" una pequeña "pieza", el llamado fibrinopéptido A y, posteriormente, el fribrinopéptido B. Esto conduce a monómeros de fibrina que inmediatamente se unen formando "polímero". Esa unión se hace más activa bajo la acción del factor XIII, estabilizando el coágulo.
Control de los mecanismos de coagulación
Frente al mecanismo hemostático natural siempre presto a dispararse para producir el coágulo, se dispone otro mecanismo complejo de función inhibidora o anticoagulante; entre ambos se procura alcanzar el equilibrio dinámico de la homeostasis sanguínea. Acción coagulante y anticoagulante se superponen en un proceso continuo que procura mantener la sangre dentro de los vasos al tiempo que asegura la permeabilidad de su luz.
Los mecanismos que actúan como inhibidores de los coagulación intravascular son varios:
- Uno de ellos es el flujo sanguíneo, que arrastra fuera del lugar de la formación del trombo sustancias procoagulantes.
- El sistema reticuloendotelial, en cuanto que elimina de la sangre circulante los factores activados de la coagulación (en el hígado, bazo y pulmón).
- Los anticoagulantes naturales conocidos como antitrombinas; han sido descritas hasta seis variedades, pero las más importantes son: la antitrombina I, que es la fibrina formada actuando como una esponja que absorbe la trombina; la antitrombina II, o cofactor de la heparina, factor plasmático necesario para la acción antitrombínica de la heparina; la antitrombina III, que lleva a cabo la neutralización de la trombina en el suero normal.
- El sistema fibrinolítico. Siendo éste el más importante componente del complejo inhibidor, conviene que le dediquemos una mayor atención.
El sistema fibrinolítico
El sistema fibrinolítoco está constituido por el plasminógeno, una pro-enzima inactiva, y aquellas sustancias que lo convierten en una forma activa, la plasmina o fibrinolisina, una enzima proteolítica responsable de la lisis de la fibrina.
Aunque el sistema fibrinolítico es el responsable de la disolución del coágulo, sus componentes también influyen en otros procesos biológicos como la ovulación, implantañción embrionaria, activación del SMF, neoplasias, reparación de tejidos y neovascularización.
El esquema de este sistema fibrinolítico se puede representar así:
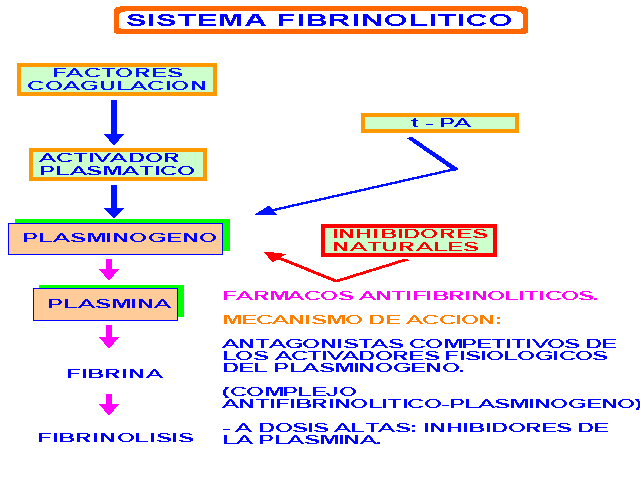 |
El plasminógeno es una proteína plasmática, pero también está presente en muchos fluídos del organismo. El lugar de síntesis es el hígado. Se convierte en plasmina por la acción de enzimas específicos llamados "activadores del plasminógeno".
Los activadores naturales del plasminógeno pueden proceder de los líquidos orgánicos, de los tejidos (activadores hísticos) o de la sangre. En estos casos se trata de proactivadores que, a su vez, son activados por otras sustancias casi siempre de origen bacteriano (estreptoquinasas, por ejemplo).
Algunos tejidos son especialmente ricos en activadores como el pulmón, cuya manipulación quirúrgica excesiva puede llegar a liberar grandes cantidades de estas sustancias potenciadoras del plasminógeno; esto explicaría la relativa frecuencia de las hemorragias postoperatorias por fibrinolisis, en la cirugía pulmonar.
En la orina se contienen también grandes cantidades de un activador muy potente al que se ha denominado uroquinasa: las hemorragias postoperatorias tras una resección transuretral de la próstata pueden ser debidas a la presencia de esta uroquinasa en la orina.
Por el contrario, el hígado contiene pocos activadores, por lo que se estima que el aumento de la actividad fibrinolítica después de una exéresis hepática sería la consecuencia de un fracaso en el mecanismo normal de eliminación por el tejido hepático de los activadores del plasminógeno.
El activador intravascular se encuentra en las paredes de los vasos sanguíneos, siendo las paredes venosas más ricas que las arteriales. Este hallazgo podría explicar por qué jlas venas se recanalizan y las arterias no. Los traumatismos vasculares reducen localmente la cantidad de activador, lo que favorecería la persistencia de las trombosis en estas zonas.
Las sustancias de procedencia bacteriana capaces de transformar los proactivadores sanguíneos en activadores completos son, fundamentalmente, la estreptoquinasa, producida por el estreptococo hemolítico y la estafiloquinasa, formada a partir del estafilococo coagulasa positivo.
A su vez, las plasminas formadas son controladas por antiplasminas naturales, que se encuentran dentro de las fracciones µ –2 y µ –1 de las globulinas. Entre los inhibidores farmacológicos de las plasminas se encuentra el ácido epsilon-aminocaproico y el trasylol, inhibidor de la kalicreína.
Bibliografía: Extraído en parte textual.
|